La diversité du règne bactérien rend son étude phylogénétique complexe : les scientifiques ne sont sont pas tous d'accord sur l'évolution qui a conduit aux espèces actuelles. De plus, certaines manières de regrouper les bactéries ont un fondement évolutif, d'autres non : mais on ne sait pas toujours lesquelles.
Pour tenter de démêler cet écheveau, nous tenterons de reconstruire la phylogénie des bactéries, à partir d'alignements multiples de séquences protéiques.
Les phylogénies obtenues seront comparées entre elles et avec celles de la littérature, afin d'estimer leur pertinence.
Sur le site Web
ftp://pbil.univ-lyon1.fr/pub/datasets/GR2002/alignments/
nous avons récupéré les séquences 3219 (preprotein translocase SecY subunit) et 11828 (30S ribosomal protein S3P).
A l'aide des premières, nous ferons une phylogénie non enracinée, à l'aide de la seconde, une phylogénie enracinée.
Comme il s'agit de séquences protéiques, nous calculons les distances à l'aide de la matrice PAM, qui tient compte de la proximité des propriétés physico-chimiques des acides aminés. Les autres méthodes disponibles sont moins pertinentes : le modèle de Poisson ne tient pas compte de ces proximités ; le nombre de différences donne une distance non additive.
Nous utilisons l'algorithme NJ (Neighbor Joining).
Le logiciel utilisé (phylowin) propose aussi BioNJ, basé sur le même principe mais moins sensible aux longues branches, donc préférable -- mais ça plante.
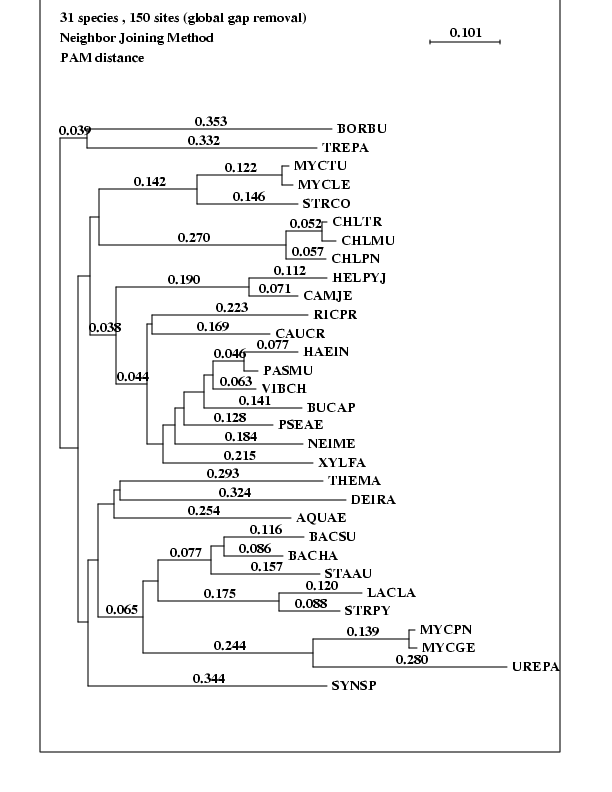
Il n'y a que des procaryotes, donc nous n'avons pas raciné l'arbre. Les différents groupes (Spirochæte, High GC gram +, Chlamydiale, Protéobactérie, Hyperthermophile, Low GC gram +) sont tous monophylétiques. Parmi les Low GC Gram +, les bacilles et les mycoplasmes forment deux groupes monophylétiques.
Pour tenter d'expliquer cette monophylie, regardons la fonction de ce gène. Pour cela, nous effectuons un Blastp (au NCBI) avec quelques unes de ces séquences ; nous avons pris celles de MYCTU (High GC gram +) et XYLFA (protéobactérie). Dans les deux cas, nous obtenons une "preprotein translocase SecY". Cette protéine est impliquée dans le transport au travers de la membrane plasmique, i.e., dans la sécrétion ; c'est une protéine transmembranaire.
Ca ne nous éclaire pas vraiment : il n'y a pas de lien clair entre la monophylie des groupes et la fonction de la protéine.
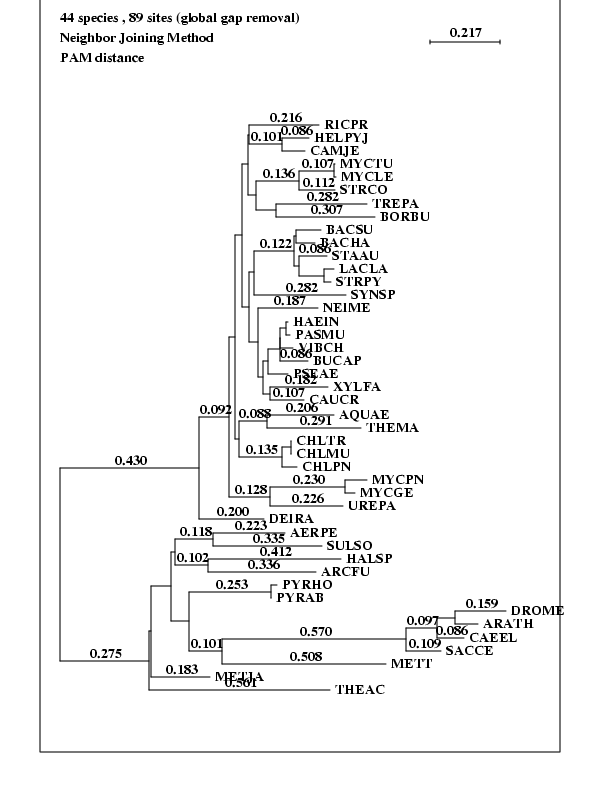
Cette fois-ci, nous avons beaucoup plus d'espèces, toujours des procaryotes, mais aussi des eucaryotes et des archaebactéries. Cela nous permet de raciner l'arbre : les procaryotes, d'une part, et les eucaryotes et les archaebactéries, d'autre part, forment deux groupes très distincts.
Les archaebactéries forment un groupe paraphylétique : leur ancètre commun est aussi un ancètre des eucaryotes.
Les archaebactéries sont en deux groupes assez distincts : d'une part, des bactéries organotrophes (plus précisément méthanotrophes, pour les bactéries étudiées), d'autre part, des bactéries chémotrophes (halophiles ou extrémophiles : elles métabolisant le soufre). Les eucaryotes semblent phylogénétiquement plus proche de ce deuxième groupe d'archaebactéries, ce qui peut paraître surprenant.
Toutefois, la branche menant aux eucaryotes est longue, le phénomène d'attraction des longues branches la rend donc peu fiable.
Contrairement à la phylogénie précédente, certains groupes sont éclatés (paraphylie) : c'est le cas des protéobactéries et des Low GC Gram +. Ces groupes restent éclatés, même si on déplace la racine (mais en fait, on n'a aucune raison de la déplacer, car on exprès ajouté des eucaryotes et des archaebactéries pour avoir un endroit privilégié où placer la racine).
Les protéobactéries G sont presque monophylétiques. Pour les rendre monophylétiques, il suffirait de retirer la protéobactérie A (CAUCR), qui a un ancètre commun avec la protéobactérie G XYLFA.
Par contre, les protéobactéries E sont monophylétiques (mais avec juste deux espèces, ce n'est pas très surprenant).
Les deux protéobactéries A (RICPR et CAUCR) sont proches du groupe des protéobactéries E pour l'une et du groupe des protéobactéries G pour l'autre.
Voici différentes hypothèses pour expliquer cet éparpillement des protéobactéries A : soit un problème avec l'algorithme utilisé (le NJ se comporte mal en présence de longues branches, or on a justement ajouté des archaebactéries et des eucaryotes par rapport à la phylogénie précédente) ; soit un problème avec le modèle évolutif (matrice PAM : il en existe d'autres, comme JTT, mais le logiciel ne les connait pas) ; soit une évolution plus rapide de cette protéine chez les protéobactéries A, de telle sorte que l'algorithme associe chacune d'elles à un groupe phylogénétique différent ; soit, la distinction entre protéobactéries A, G et E est peu pertinente, au moins pour la protéine étudiée.
Les Low GC Gram + sont réparties en deux groupes monophylétiques : d'une part les bacilles, d'autre part les mycoplasmes. Il est possible que les Low GC Gram + ne forment pas un groupe monophylétique ; en effet, la similarité entre les pourcentage de GC est peut-être le résultat d'une convergence ; par contre, la monophylie des bacilles et des mycoplasmes a une réalité évolutive : les bacilles ont une paroi en plus de la membrane, contrairement aux mycoplasmes qui n'ont qu'une membrane -- c'est cohérent avec leur position dans l'arbre : le groupe des mycoplasmes semble avoir peu évolué depuis l'ancètre commun.
Pour critiquer ces résultats, nous allons les comparer avec d'autres phylogénies des bactéries : d'une part, une phylogénie calculée de la même manière avec plusieurs de ces protéines, d'autre part, la phylogénie de l'article dont viennent ces données, enfin, d'autres phylogénies, provenant de la littérature.

On distingue différents groupes (de haut en bas, sur le dessin) : la cyanobactérie, les High GC Gram +, les hyperthermophiles, les bacilles, les mycoplasmes, les chlamidiae, les protéobactéries E, les spirochètes, les protéobactéries B, G, A. Chacun de ces groupes est monophylétique.
La seule différence avec notre première analyse phylogénétique, c'est l'éclatement des protéobactéries : la protéine que nous avions prise est probablement soumise à une pression évolutive élevée (protéine membranaire chez des extrémophiles), alors que certaines ont pu évoluer et diverger plus librement.
A part ce détail, notre première analyse phylogénétique apportait la même information.
A phylogenetic approach to bacterial phylogeny: evidence of a core of genes sharing a commun history http://www.ncbi.nlm.nih.gov:80/entrez/utils/fref.fcgi?http://www.genome.org/cgi/pmidlookup?view=full&pmid=12097345
Ils rappellent que la prise en compte des transferts horizontaux change radicalement la topologie de l'"arbre" et met en évidence des phénomènes de "paralogie cachée" (xénologie) ; et qu'ils convient de retirer les longues branches qui nuisent à la précision de l'algorithme.
Ils constatent que le maximum de parcimonie produit des artéfacts, confirmant le fait que cette méthode est peu adaptée à la reconstruction d'une phylogénie à partir de séquences protéiques. Comme nous avons utilisé une distance calculée à partir de la matrice PAM, nous n'avons pas constaté ces artéfacts.
La présence des mycoplasmes et des bacilles dans un même groupe monophylétique confirme la robustesse de leur approche. Nous avons les mêmes résultats, sauf pour l'arbre contenant aussi des eucaryotes et des archaebactéries : les longues branches qu'ils entrainent perturbent l'algorithme. Idéalement, il faudrait construire la phylogénie des procaryotes séparément et, une fois cette pylogénie non racinée construite, ajouter eucaryotes et archaebactéries afin de racine l'arbre -- mais le logiciel utilisé ne le permet pas.
La variabilité des taux d'évolution, i.e., la présence de longues branches, influence l'arbre : les espèces évoluant très vite ont une position basale. Leur algorithme est robuste face à ce genre de problèmes ; les nôtres aussi.
La position de Deinococcus et des thermophiles évoque une possible polyphylie chez les High GC Gram + : certaines de nos phylogénies confirment cela, mais pas toutes. Pour élucider la phylogénie de ce groupe, l'article suggère de regarder des données plus précises, comme l'évolution de la DNA polymérase C.
Globalement, nos résultats sont comparables aux leurs.
Comparons nos résultats avec ceux de :
Phylogénie des eubactéries basée sur l'ARNr 16S Université de Provence, Marseille http://www.up.univ-mrs.fr/evol/phylogenomics-lab/celine/cours2.pdf http://www.up.univ-mrs.fr/evol/phylogenomics-lab/celine/cours2.ppt
Le fichier n'est plus là, mais vous pouvez (peut-être) consulter la copie que Google en a fait (il manque toutefois le plus important : les dessins) :
http://www.google.fr/search?q=cache:rw15pXXzf6cJ:www.up.univ-mrs.fr/evol/phylogenomics-lab/celine/cours2.pdf+++Phylog%C3%A9nie+des+eubact%C3%A9ries+bas%C3%A9e+sur+l%27ARNr+16S%0D%0A&hl=fr&ie=UTF-8
Cette phylogénie comporte des rateaux (correspondant à des arêtes qui n'apparaissent que rarement lorsqu'on lance l'algorithme sur plusieurs échantillons de bootstrap), contrairement aux nôtres, car le logiciel utilisé ne le permet pas.
Les protéobactéries sont regroupées, comme dans nos phylogénies : les protéobactéries A, B, E, G dont monophylétiques, mais pas les protéobactéries D : nous n'en avons pas, il aurait été intéressant de voir où elles se placent.
Les spirochètes et les hyperthermophyles sont des groupes monophylétiques, comme dans nos phylogénies.
Les High GC forment un groupe monophymétique, mais on ne peut pas résoudre les Low GC : c'est conforme avec nos reconstructions, dans lesquelles les Low GC forment deux groupes (mycoplasmes et bacilles).
La comparaison avec d'autres phylogénies donnerait des résultats comparables.
Nos reconstructions phylogénétiques (notre première phylogénie) ne sont pas liées au mode de vie (libre ou parasitaire) des bactéries
Spirochètes, Chlamidiae et Low GC sont pathogènes (le plus souvent parasites humains) ; les protéobactéries, High GC, hyperthermophiles et cyanobactéries occupent des niches écologiques hostiles. Cependant, ces deux groupes, pathogènes et extrémophiles, ne sont pas monophylétiques : le mode de vie est un caractère polyphylétique.
Voici une explication possible : il y a beaucoup d'environements extrèmes différents, les modes de vie parasitaires sont eux aussi très variés. Il a avait donc une forte probabilité pour qu'une espèce s'y adapte.
La figure suivante est extraite de
http://www.sci.sdsu.edu/~smaloy/MicrobialGenetics/topics/chroms-genes-prots/genomes.html
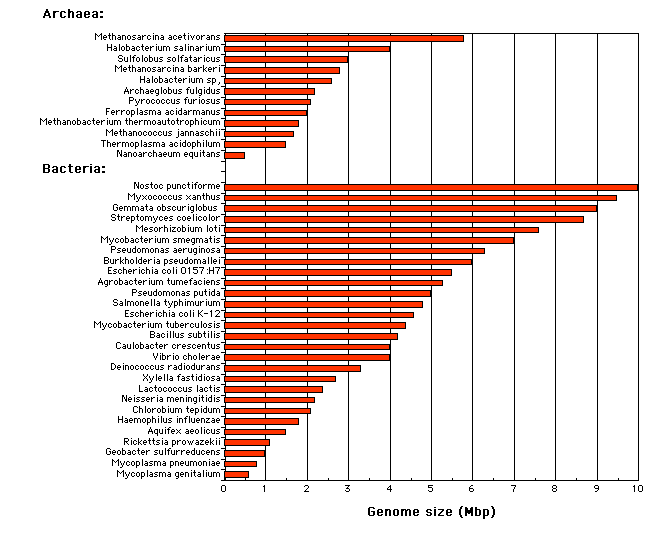
Les mycoplasmes ont un petit génome ; mais les autres groupes sont très hétérogènes, ainsi les protéobactéries ont souvent un génome de taille moyenne, mais certains sont petits (Rickttsia), d'autres grands (Pseudomonas).
Comme le mode de vie, la taille du gènome est généralement un caractère polyphylétique. Il n'y a pas de lien direct entre la taille du gènome et la morphologie.
Voici une explication possible : le transfert horizontal de gènes peut faire passer un gène d'un petit génome vers un gros génome ou l'inverse, d'où le mélange des tailles.
Les protéines choisies,
3219 : Preprotein translocase SecY subunit 11828 : 30S ribosomal protein S3P
sont présentes chez les procaryotes étudiés (la seconde est même vitale) et présentent une diversité suffisemment grande pour effectuer une analyse phylogénétique.
Les espèces choisies sont elles aussi variées, permettant d'avoir une vue assez complète de la phylogénie des procaryotes.
Les résultats sont dépendants de la protéine étudiée. Néanmoins, certains groupes monophylétiques demeurent monophylétiques dans toutes les phylogénies considérées.
Les algorithmes et logiciels utilisés sont toujours insuffisants : ils restent très sensibles au phénomène d'attraction des longues branches et ne tiennent pas compte du transfert horizontal de gènes, malgré son importance chez les procaryotes.
Olivier Poirot, Vincent Zoonekynd
latest modification on Wed Mar 17 16:06:22 CET 2004